
Pédagogie Numérique en médecine 🎭 Les cafés numériques de la santé publique
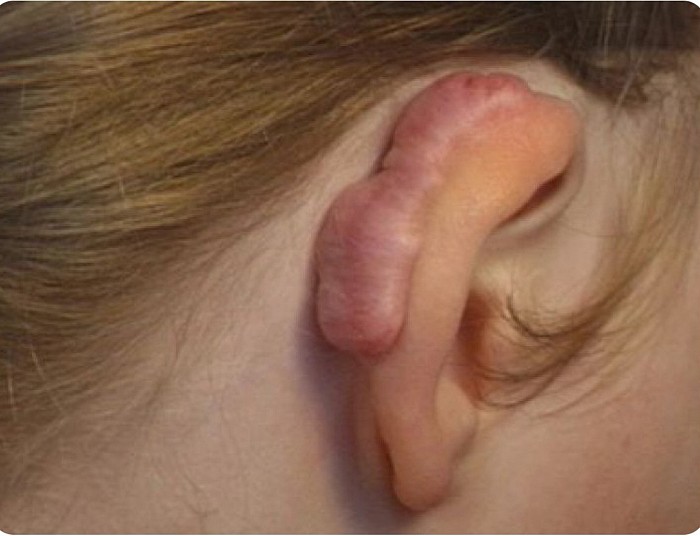
Chéloïde
Chéloïde
Chéloïde..
L'excision entraînera une récidive dans la plupart des cas.
Les injections intralésionnelles de stéroïdes, la cryothérapie, les pansements en silicone peuvent aider à améliorer.
Même si cela ne disparaîtra pas complètement chez une personne prédisposée.
Syndrome de Cushing
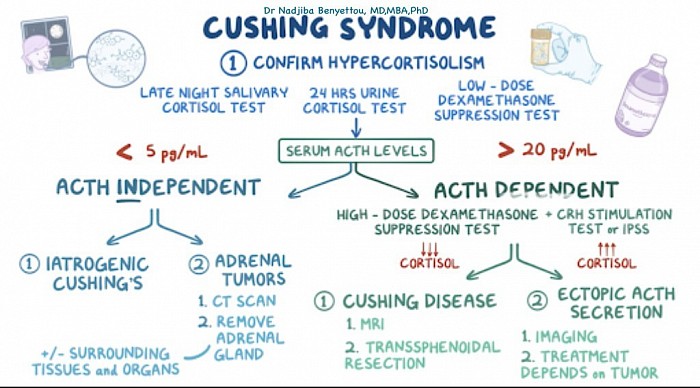
Syndrome de cushing
À retenir
Le bilan de santé de la maladie de Cushing consiste d’abord à vérifier les niveaux élevés de cortisol. Chez les patients présentant des taux de cortisol élevés, l’étape suivante consiste à mesurer l’ACTH plasmatique. Parmi les patients présentant un taux d'ACTH élevé, un test à la dexaméthasone à haute dose peut distinguer un adénome hypophysaire d'une sécrétion ectopique d'ACTH. La résection chirurgicale est le traitement de choix de la maladie de Cushing.
Explication principale
Les résultats cliniques et biologiques chez ce patient sont cohérents avec une maladie de Cushing secondaire à une surproduction d'ACTH par un adénome hypophysaire. Ces patients peuvent développer une faiblesse musculaire, une prise de poids, un amincissement de la peau, des ecchymoses et une redistribution des graisses. De plus, les patients peuvent développer une hyperglycémie secondaire à une tolérance réduite au glucose.
La première étape du bilan consiste à mesurer le cortisol libre dans un échantillon d’urine de 24 heures. Les méthodes alternatives comprennent des analyses de sang ou de salive et un test de suppression de la dexaméthasone à faible dose.
Si l’un des tests susmentionnés donne des résultats anormaux, l’étape suivante consiste à déterminer la cause exacte de la production excessive de cortisol. Cette évaluation consiste à mesurer les taux plasmatiques d'ACTH. De faibles taux d'ACTH sont observés dans les adénomes surrénaliens, les carcinomes surrénaliens et l'utilisation de glucocorticoïdes exogènes. Des taux élevés d'ACTH sont observés chez les patients atteints de la maladie de Cushing (adénome hypophysaire sécrétant de l'ACTH) et d'une production ectopique d'ACTH.
Chez les patients présentant des taux d’ACTH élevés, l’étape suivante consiste à administrer une injection à forte dose de dexaméthasone. S’il n’y a pas de baisse des niveaux d’ACTH/cortisol, cela signifie qu’il existe un site ectopique de production d’ACTH. Si les taux d’ACTH/cortisol chutent après le test à la dexaméthasone à haute dose, cela signifie qu’un adénome hypophysaire sécrétant de l’ACTH en est la cause.
Le patient de cette vignette présente des taux d'ACTH élevés et une production endogène réduite de cortisol après l'administration de fortes doses de dexaméthasone, ce qui est compatible avec la maladie de Cushing. La résection chirurgicale constitue l’option thérapeutique de première intention.
Thromboembolie
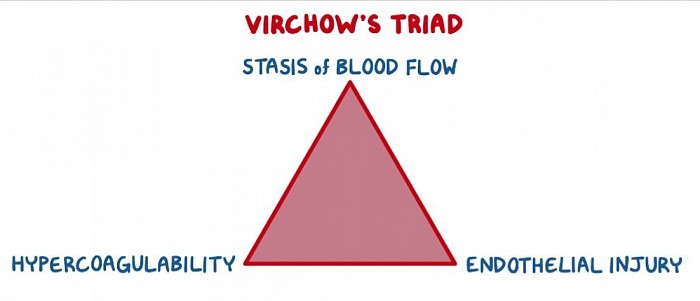
Thrombose
À retenir
Les facteurs de risque de développement d'une thromboembolie veineuse sont résumés par les facteurs qui contribuent à la triade de Virchow : stase veineuse, lésion endothéliale et état d'hypercoagulabilité. Les facteurs de risque prédisposant les plus importants au développement d'une thromboembolie veineuse sont une intervention chirurgicale majeure récente, des troubles thrombophiliques héréditaires, des antécédents de TVP, un âge > 70 ans, une grossesse et un cancer actif.
Explication principale
Ce patient souffre probablement d’une thrombose veineuse profonde (TVP), qui se présente classiquement comme un membre inférieur unilatéral, enflé et douloureux. La triade de Virchow résume la physiopathologie conduisant à la thromboembolie veineuse : lésion endothéliale, hypercoagulabilité et stase veineuse.
Les lésions endothéliales peuvent résulter d'une inflammation chronique ou d'un traumatisme vasculaire, comme chez les patients cancéreux, d'une infection ou d'un traumatisme/chirurgie. L'hypercoagulabilité peut être congénitale (par exemple, facteur V Leiden), acquise (par exemple, syndrome néphrotique) ou due à des médicaments qui induisent une hypercoagulabilité comme un traitement hormonal substitutif. La stase veineuse survient pendant les périodes d'immobilité prolongée, notamment les vols en avion, les séjours à l'hôpital, les opérations postopératoires ou les modes de vie sédentaires.
Parmi tous les facteurs de risque présentés par ce patient, une intervention chirurgicale majeure récente est le facteur de risque le plus important avec un risque relatif de 5 à 200. D'autres facteurs de risque majeurs conférant un risque relatif élevé comprennent les troubles thrombophiliques héréditaires, les antécédents de TVP, l'âge > 70 ans et la grossesse. D'autres facteurs de risque connus incluent l'obésité et le traitement de remplacement des œstrogènes, mais ceux-ci comportent un risque relatif plus faible de 1 à 3.
Tous les principaux facteurs de risque de TVP peuvent être résumés à l’aide du mnémonique « THROMBOSE » :
BRAF ET Melanoma
À retenir
La mutation génétique la plus courante observée dans le mélanome implique BRAF, une protéine kinase impliquée dans l'activation des voies de signalisation de la prolifération des mélanocytes. Le vémurafénib est un anticorps monoclonal dirigé contre la BRAF kinase, bloquant ainsi la prolifération cellulaire dans les cellules cancéreuses.
Explication principale
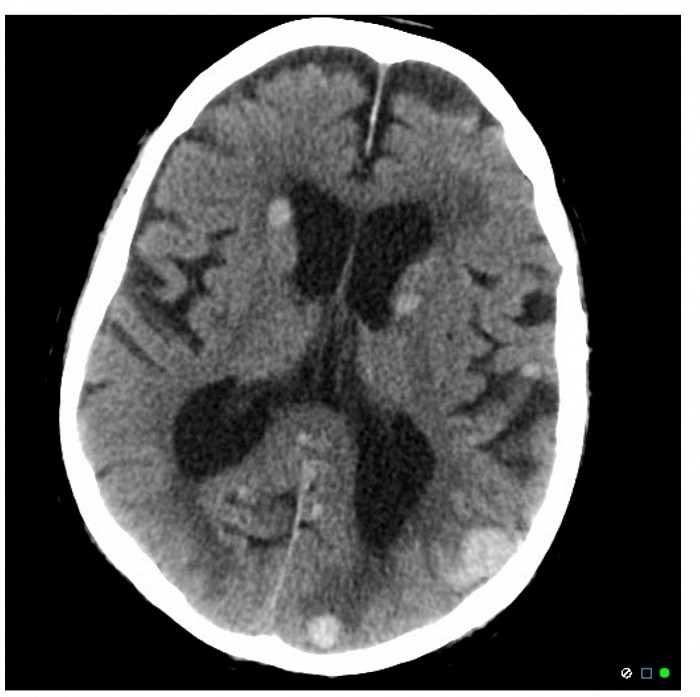
La présentation de ce patient avec des signes et des symptômes de lésions occupant de l'espace (par exemple, déficits neurologiques focaux, convulsions) et des preuves d'imagerie tomodensitométrique de multiples lésions rondes sont fortement évocatrices d'une maladie métastatique. Dans ce cas, il s’agit probablement d’un mélanome, une affection dermatologique qui répond généralement bien au vémurafénib.
Le mélanome est la forme la plus agressive de tumeur maligne cutanée avec des taux élevés de métastases. La mutation génétique la plus courante observée dans les tissus néoplasiques concerne BRAF (jusqu'à 60 % des cas), qui est une protéine kinase impliquée dans l'activation des voies de signalisation de la prolifération des mélanocytes. Sa mutation ponctuelle entraîne une substitution de la valine par l'acide glutamique en position 600 de la protéine. Le résultat final est une activation continue de BRAF, qui provoque une prolifération incontrôlée de mélanocytes atypiques. Le vémurafénib est un anticorps monoclonal dirigé contre la BRAF kinase, bloquant ainsi la prolifération cellulaire dans les cellules cancéreuses. C'est un agent privilégié chez les patients atteints de mélanome métastatique dont le test est positif pour la mutation BRAF V600E.
Les (TdT) et le CD10 sont les précurseurs de quels types de cellules immunitaires ?
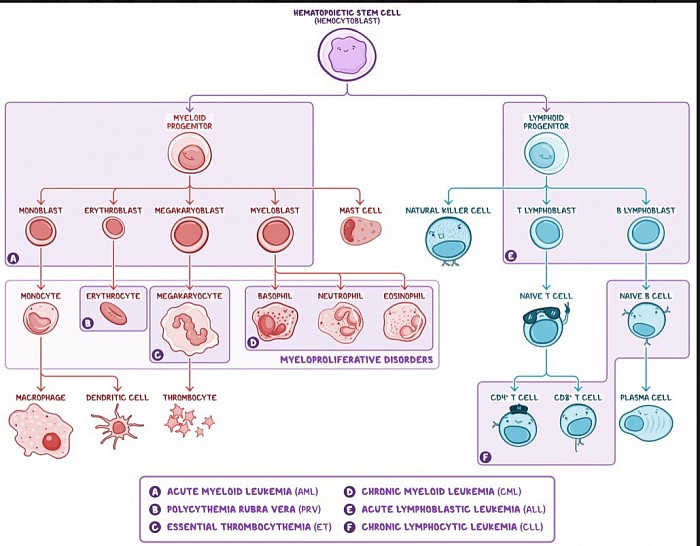
CD10 et TdT
Bien que les précurseurs des lymphocytes T et B semblent similaires sur les frottis sanguins périphériques et qu'ils soient tous deux positifs pour le TdT, les marqueurs de surface des lymphocytes pré-T sont différents et incluent CD2-5, CD7 et CD8. De plus, la LAL-T se présente généralement à l'adolescence avec une masse médiastinale antérieure et/ou des effets de masse (p. ex., syndrome de type SVC, dysphagie, dyspnée).
À retenir
La LAL est due à des mutations dans NOTCH1, t(12;21) ou, rarement, t(9;22). Elle se manifeste par une pancytopénie, une hépatosplénomégalie, une lymphadénopathie et/ou des effets de masse. Les lymphoblastes sont caractérisés par une coloration nucléaire positive pour le TdT. La biopsie de la moelle osseuse montre ≥20 % de lymphoblastes et le diagnostic est confirmé par cytométrie en flux.
Explication principale
La présentation de cet enfant avec de la fièvre, de la fatigue et des maux de gorge (infection), ainsi que des lymphoblastes sur le frottis périphérique et une coloration cellulaire positive pour la désoxynucléotidyl transférase terminale (TdT) et le CD10, suggèrent fortement une leucémie lymphoblastique aiguë à cellules B (LAL-B).
La LAL est une tumeur des cellules pré-B ou pré-T. Elle est définie comme l’expansion clonale de lymphoblastes malins dans la moelle osseuse. C'est le cancer le plus répandu chez les enfants et il survient plus fréquemment chez les garçons. Il est associé au syndrome de Down. La pathogenèse implique des aberrations chromosomiques, notamment des mutations de NOTCH1 (T-ALL) ou une translocation de t(12;21) ou, rarement, de t(9;22) (B-ALL). La LAL-B représente environ 70 à 80 % des cas et culmine vers l'âge de 3 ans. Les lymphoblastes sont positifs pour la TdT, une ADN polymérase spécialisée exprimée dans les cellules pré-B et pré-T. Les lymphoblastes B expriment CD10, CD19 et CD20.
Cliniquement, la LAL se manifeste par des signes d'anémie (fatigue, dyspnée), de thrombocytopénie (ecchymoses faciles), de leucocytes dysfonctionnels (infections), d'hépatosplénomégalie et/ou de lymphadénopathie. Elle peut également se manifester par des effets de masse secondaires à une infiltration néoplasique du sous-périoste (douleurs osseuses, refus de supporter du poids), du système nerveux central (maux de tête, vomissements) ou, plus rarement, des testicules (observé dans la LAL-T). La biopsie de la moelle osseuse montre ≥20 % de lymphoblastes dotés d'un cytoplasme basophile peu abondant et d'un gros noyau. Le diagnostic définitif nécessite la preuve immunophénotypique de la lignée lymphoïde par cytométrie en flux. Le pronostic dépend de l'âge, du sexe et de la translocation (t[9;22] est associé à un pire pronostic).
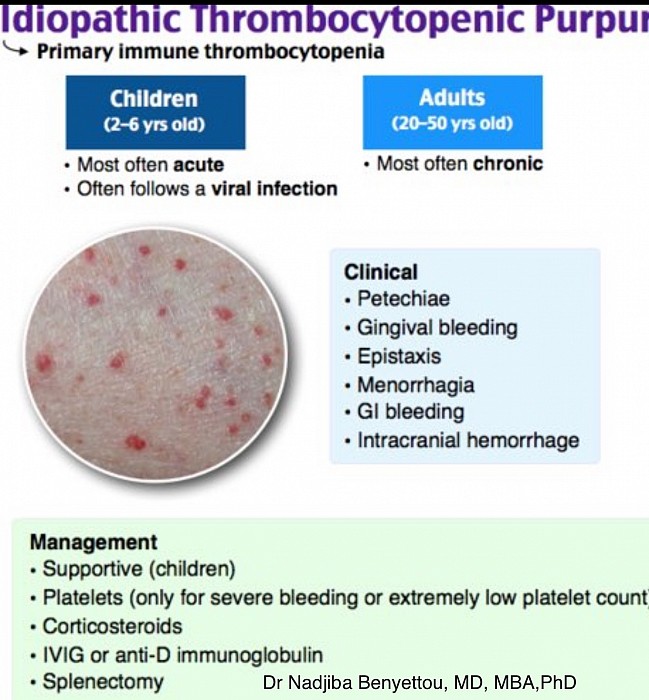
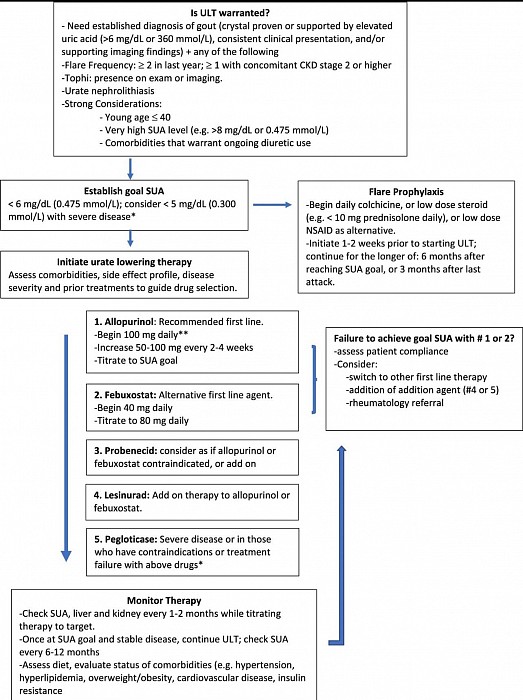
La Goutte
Les porphyries
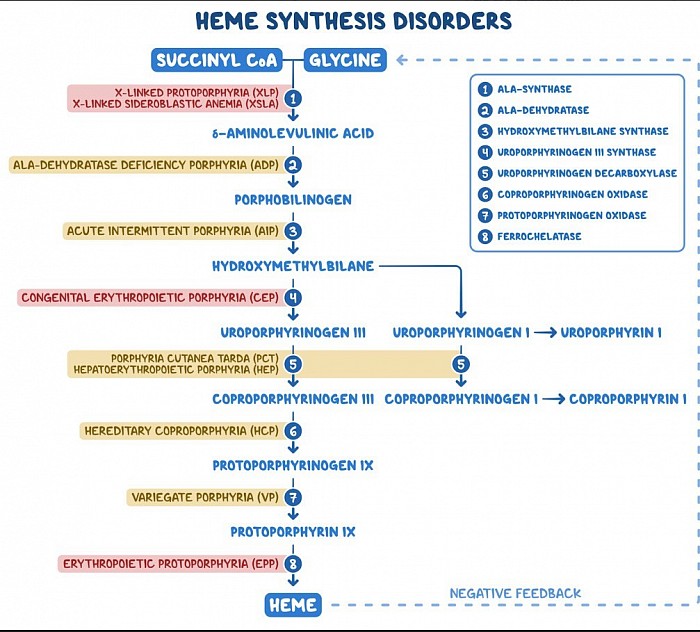
À retenir
Les porphyries sont des troubles métaboliques causés par une altération des activités des enzymes au sein de la voie de biosynthèse de l'hème. Ces conditions conduisent à l’accumulation de précurseurs de l’hème. Des exemples de porphyries comprennent la porphyrie cutanée tardive (la plus courante), la porphyrie aiguë intermittente, la porphyrie par déficit en acide aminolévulinique déshydratase et la coproporphyrie héréditaire.
Explication principale
Ce patient présente des douleurs abdominales, des troubles psychiatriques, des paresthésies et des urines brun rougeâtre. De plus, elle a eu des symptômes similaires dans le passé qui se sont ensuite résolus d’eux-mêmes. Elle souffre probablement de porphyrie aiguë intermittente (AIP), causée par des défauts de la porphobilinogène désaminase.
Les porphyries sont des troubles métaboliques causés par une altération des activités des enzymes au sein de la voie de biosynthèse de l'hème. Ces conditions conduisent à l’accumulation de précurseurs de l’hème. La plupart des porphyries sont héréditaires, à l'exception de la porphyrie cutanée tardive (PCT), qui est généralement acquise. Les porphyries sont classées comme hépatiques ou érythropoïétiques selon que les produits héminiques s'accumulent d'abord dans le foie ou la moelle osseuse.
Les porphyries hépatiques sont généralement présentes chez les adultes présentant des symptômes neurologiques, notamment des douleurs abdominales neuropathiques, une neuropathie motrice périphérique et des troubles mentaux. En revanche, les porphyries érythropoïétiques se présentent généralement peu de temps après la naissance ou dans la petite enfance avec une photosensibilité cutanée.
Les différents types de porphyries comprennent la porphyrie cutanée tardive (la plus courante), la porphyrie aiguë intermittente, la porphyrie par déficit en acide aminolévulinique déshydratase, la coproporphyrie héréditaire, la porphyrie panachée et la porphyrie érythropoïétique congénitale.
Hypertension artérielle
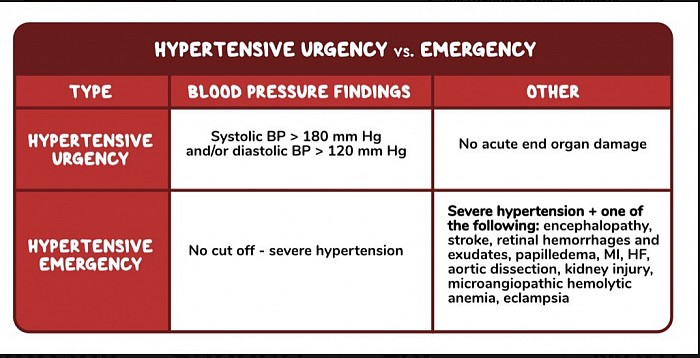
À retenir
L'urgence hypertensive est définie comme une pression artérielle très élevée (généralement supérieure à ≥180/≥120) avec des signes de lésions des organes cibles. Les patients peuvent présenter des maux de tête, de la confusion, des nausées et des vomissements ainsi qu'une vision floue. Ce diagnostic diffère de l’urgence hypertensive, qui correspond à une pression artérielle très élevée sans aucun signe de lésion des organes cibles.
Explication principale
Ce patient ayant des antécédents d'hypertension et qui présente maintenant des maux de tête d'apparition soudaine, une pression artérielle extrêmement élevée, une vision floue, de la confusion et un œdème pulmonaire est très probablement en situation d'urgence hypertensive.
Lorsque l’élévation de la pression artérielle atteint un niveau critique (généralement autour de 180/120 mm Hg), la réponse myogénique vasculaire augmente la résistance vasculaire. En réponse à une augmentation de la pression intravasculaire, la réponse myogénique est une vasoconstriction vasculaire. L'hypoperfusion périphérique relative provoque une augmentation des hormones vasoactives, telles que l'angiotensine II, la noradrénaline et l'hormone antidiurétique ; cette réponse inadaptée aggrave la résistance périphérique et conduit à un cercle vicieux d’aggravation de l’hypertension et de lésions des organes. Dans ce cas d’urgence hypertensive, l’œdème pulmonaire du patient est très probablement causé par un dysfonctionnement ventriculaire gauche dû à une hypertension artérielle, entraînant un reflux de sang dans le système vasculaire pulmonaire.
L'urgence hypertensive est définie comme une pression artérielle systolique et diastolique supérieure à ≥180/≥120 mmHg respectivement sans lésion aiguë d'un organe terminal. En revanche, une urgence hypertensive est définie comme une pression artérielle très élevée accompagnée de signes de lésions aiguës des organes cibles. Ceux-ci inclus:
Neurologique : encéphalopathie, accident vasculaire cérébral, hémorragies et exsudats rétiniens, œdème papillaire
Cardiopulmonaire : Infarctus du myocarde, insuffisance cardiaque avec œdème pulmonaire
Vasculaire : dissection aortique, anémie hémolytique microangiopathique
Rénal : néphrosclérose hypertensive aiguë
Le diagnostic repose sur les antécédents et l'examen physique des patients présentant une tension artérielle significativement élevée, qu'ils aient ou non des antécédents d'hypertension. Des tests de diagnostic supplémentaires doivent être effectués pour évaluer la présence de lésions d'un organe cible, notamment un ECG, une analyse d'urine, une radiographie pulmonaire, des électrolytes sériques, de la créatinine sérique, des biomarqueurs cardiaques et une imagerie de la tête. Le traitement consiste en une réduction pharmacologique de la pression artérielle (par exemple, nitroprussiate, hydralazine) ; cependant, cela doit être fait progressivement pour éviter les complications ischémiques.
L'échelle de Tanner
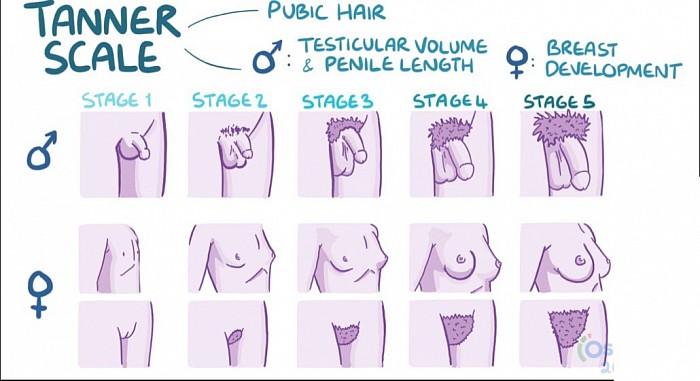
À retenir
Le stade Tanner 2 chez les mâles est caractérisé par l’apparition de poils pubiens mous et le début d’une hypertrophie testiculaire.
Explication principale
L'échelle de Tanner est une échelle utilisée pour évaluer le développement des caractéristiques sexuelles primaires et secondaires chez les hommes et les femmes, et elle représente un ensemble d'étapes par lesquelles passent les hommes et les femmes à la puberté. L'échelle Tanner évalue trois critères indépendants : les organes génitaux, les poils pubiens et le développement mammaire chez la femme. Il y a cinq étapes dans l’échelle du tanneur :
Stade 1 : le stade prépubère ; aucun poil pubien n'est présent ni chez les hommes ni chez les femmes ; les mâles ont un petit pénis et des testicules ; les femelles ont une poitrine plate.
Stade 2 : des poils pubiens mous commencent à apparaître chez les hommes et les femmes ; les testicules commencent à grossir chez les mâles ; des bourgeons mammaires apparaissent chez les femelles.
Stade 3 : les poils pubiens commencent à grossir ; le pénis commence à grossir en taille et en longueur ; chez les femelles, des monticules mammaires se forment et les seins grossissent.
Stade 4 : les poils pubiens recouvrent la zone pubienne ; le pénis s'élargit ; les seins continuent de grossir et forment un monticule sur le contour du monticule.
Stade 5 : les poils pubiens s’étendent jusqu’à l’intérieur des cuisses ; le pénis et les testicules atteignent la taille adulte ; les seins atteignent le contour adulte.
La dysplasie fibreuse des os (DFO)
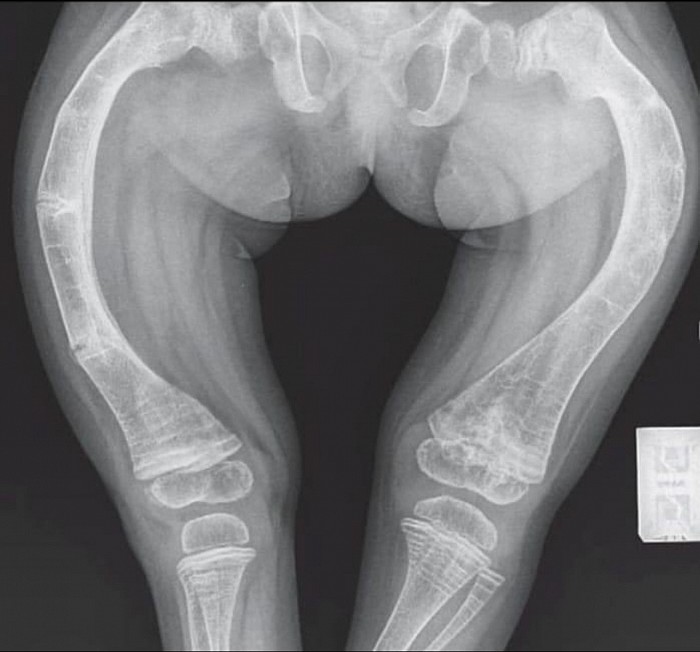
La dysplasie fibreuse des os (DFO)
La dysplasie fibreuse des os (DFO) est une maladie caractérisée par une anomalie de cer- tains os qui peuvent devenir fragiles et se déformer. N'importe quel os peut être touché, mais il s'agit souvent du fémur (os de la cuisse), du tibia (os de la jambe), des côtes, des os du crâne. Elle entraîne parfois des douleurs au niveau du ou des os atteint(s), peut augmenter le risque de fracture et entraîner des déformations osseuses.
Des complications neurologiques peuvent être associées lorsque les os du crâne sont at- teints et il peut, exceptionnellement, y avoir des complications hormonales.
l Combien de personnes sont atteintes de la maladie ?
C’est une maladie rare, mais sa prévalence (nombre de cas dans une population donnée à un moment précis) est difficile à estimer car, souvent, la maladie n’entraîne aucun symptôme.
l Qui peut en être atteint ? Est-elle présente partout dans le monde ?
Cette maladie atteint aussi bien les filles que les garçons, quelle que soit leur origine géo- graphique. L’âge au moment du diagnostic est le plus souvent compris entre 5 et 30 ans.
l Est-elle contagieuse ? Non, la DFO n’est pas contagieuse.
l A quoi est-elle due ?
La DFO est due à une anomalie génétique, c’est-à-dire qu’elle est provoquée par l’altération
(mutation) d’un gène appelé GNAS dans certaines cellules du corps (des os, de la peau,...).

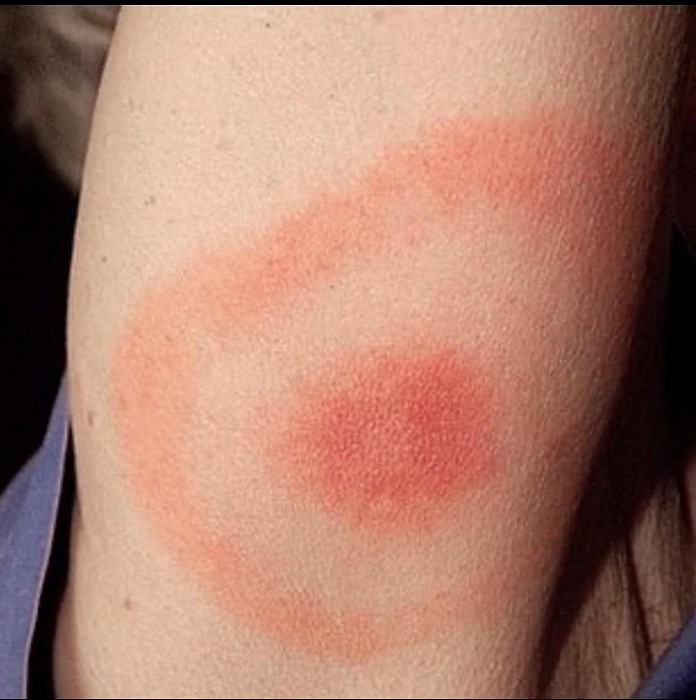
Lyme disease
